Moustafa Hadi, BS1, Rami M. Youssef, MD2, Mohsen Dourra, MD3, Mark Obri, MD2, Mohamad Beidoun, MD4, Syed-Mohammed Jafri, MD5 1Michigan State University College of Human Medicine, Detroit, MI; 2Henry Ford Hospital, Detroit, MI; 3Corewell Health, Woodhaven, MI; 4Henry Ford Hospital, Livonia, MI; 5Henry Ford Health System, Detroit, MI
Introduction: Existing on a spectrum of congenital biliary cystic diseases, Caroli syndrome (CS) is a rare autosomal recessive condition exhibiting non-obstructive segmental intrahepatic biliary duct dilation concurrent with hepatic fibrosis. We present a patient presenting with abdominal pain and elevated liver enzymes diagnosed as CS.
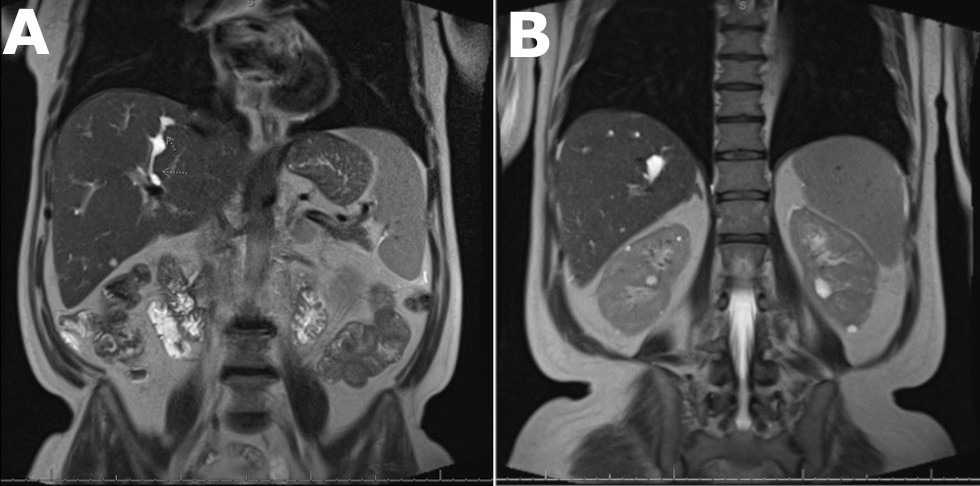
Case Description/Methods: A 57-year-old female presents with chronic right upper quadrant (RUQ) abdominal pain, nausea, and chills. She has a history of recurrent cholangitis and cholecystectomy. Examination reveals hepatomegaly with mild pain to palpation. Blood cultures are growing Enterococcus faecium. Workup demonstrates elevated alkaline phosphatase (313 IU/ml), gamma-glutamyl transferase (203 IU/L), C-Reactive Protein (85.3 mg/dL), and erythrocyte sedimentation rate (120 mm/hr). Total bilirubin is within normal limits (0.2 mg/dL). Wedge liver biopsy revealed congenital hepatic fibrosis with associated bile duct proliferation and obstruction. Magnetic resonance cholangiopancreatography (MRCP) revealed mild liver surface nodularity, multifocal saccular dilatation of the right posterior intrahepatic bile ducts with scattered bilobar biliary stricturing with no dominant stricture [Figure A], and several small renal cysts [Figure B]. Based on imaging, biopsy, and history, the patient was diagnosed with CS. Her Model for End-Stage Liver Disease (MELD) score is 7. Repeat blood cultures were negative and her symptoms improved with antibiotics.
Discussion: Although the prevalence ranges from 1 in 1,000,000 to 1 in 100,000, CS should be considered in a patient with recurrent RUQ abdominal pain and cholangitis. Presentation may also be complicated by hepatic abscess and sepsis. Prompt diagnosis is paramount to prevent and monitor for progressive hepatic injury, portal vein thrombosis, cholangiocarcinoma, and renal disease. Those with CS have a 100-fold increased risk of developing malignancy. Autosomal recessive polycystic kidney disease is associated with CS as both conditions are related to a mutation of the PKHD1 gene. MRCP differentiates CS from similar diseases such as primary sclerosing cholangitis (PSC). The morphology of CS is segmental and saccular whereas that of PSC is isolated and fusiform. Management depends on the degree of hepatic fibrosis and biliary tract involvement. Definitive therapy includes lobular resection in local disease or OLT in diffuse disease with fibrosis. The patient is currently discussing liver transplantation due to CS with cholangitis.
Figure: Figure A: Magnetic resonance imaging of the abdomen with contrast showing saccular dilatation of multiple biliary radicles within the posterior right lobe. Figure B: Magnetic resonance imaging of the abdomen with contrast demonstrating renal cysts.
Disclosures:
Moustafa Hadi indicated no relevant financial relationships.
Rami Youssef indicated no relevant financial relationships.
Mohsen Dourra indicated no relevant financial relationships.
Mark Obri indicated no relevant financial relationships.
Mohamad Beidoun indicated no relevant financial relationships.
Moustafa Hadi, BS1, Rami M. Youssef, MD2, Mohsen Dourra, MD3, Mark Obri, MD2, Mohamad Beidoun, MD4, Syed-Mohammed Jafri, MD5. P1544 - A Late Diagnosis of Caroli Syndrome, ACG 2023 Annual Scientific Meeting Abstracts. Vancouver, BC, Canada: American College of Gastroenterology.