Jennifer Dimino, MD1, Joanna Lenik, MD2, Maria Camila Fonseca, MD3, Maryam Banikazemi, MD4, Nora V. Bergasa, MD5 1NYCHH/Metropolitan Hospital, New York, NY; 2NYC Health + Hospitals/Metropolitan Hospital, Manhattan, NY; 3New York Health and Hospitals/Woodhull Hospital, Brooklyn, NY; 4Health + Hospitals/Metropolitan Hospital Center, Manhattan, NY; 5Health + Hospitals/Woodhull Medical Center, Brooklyn, NY
Introduction: Isolated polycystic liver disease (IPLD) is a rare genetic disorder present in 0.01% to 1% of Nordics with polycystic kidney disease (PCKD)[1][2]. However, no reports have been published on patients of African descent. We present a case of an African woman diagnosed with IPLD by genetic testing.
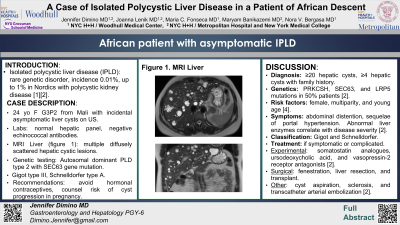
Case Description/Methods: A 24-year-old woman (G3P2) from Mali was referred to the Hepatology clinic for evaluation of liver cysts incidentally found by ultrasound done for low abdominal pain, not secondary to the cysts. The hepatic panel was normal and echinococcal antibodies were negative. A liver MRI showed multiple diffusely scattered cystic lesions throughout the liver, with a subcentimeter left renal cyst. Genetic testing revealed autosomal dominant PLD type 2 with a pathogenic variant in the SEC63 gene. Recommended to avoid hormonal contraceptives, and be aware of the higher risk of cyst progression if becoming pregnant.
Discussion: IPLD is defined as ≥20 hepatic cysts (or ≥4 with family history) and 50% of the patients have recognized gene mutations (PRKCSH, SEC63 and LRP5) [2]. Risk factors include female gender, multiparity, and young age [4]. Symptoms vary from abdominal distention or pain to sequelae of portal hypertension. Abnormal liver enzymes correlate with disease severity [2]. Two clinical classifications exist; Gigot, types I to III, which includes the number and size of cysts, and the remaining area of non-cystic parenchyma, and Schnelldorfer types A to D, which includes symptoms, cyst characteristics, area of normal parenchyma and isosectoral portal or hepatic vein occlusion. The patient was classified as Gigot type III; Schnelldorfer type A [2]. Only symptomatic or complicated cysts require intervention. There are no approved drugs for IPLD. Experimental pharmacologic treatments include somatostatin analogs, ursodeoxycholic acid, and vasopressin-2 receptor antagonists [2]. Other therapies include cyst aspiration, sclerosis and transcatheter arterial embolization [2]. Surgical therapies include fenestration, liver resection, and transplant. The therapeutic alternative is liver transplantation for patients with severe symptoms and significant decline in quality of life with portal hypertension or malnutrition [2]. Invasive interventions should be done by experienced multidisciplinary teams, based on the patients’ clinical classification and Child-Pugh score. Studies on disease burden, symptom assessment, and response to proposed therapies are warranted.
Disclosures:
Jennifer Dimino indicated no relevant financial relationships.
Joanna Lenik indicated no relevant financial relationships.
Maria Camila Fonseca indicated no relevant financial relationships.
Maryam Banikazemi indicated no relevant financial relationships.
Nora V. Bergasa indicated no relevant financial relationships.
Jennifer Dimino, MD1, Joanna Lenik, MD2, Maria Camila Fonseca, MD3, Maryam Banikazemi, MD4, Nora V. Bergasa, MD5. P2484 - A Case of Isolated Polycystic Liver Disease in a Patient of African Descent, ACG 2023 Annual Scientific Meeting Abstracts. Vancouver, BC, Canada: American College of Gastroenterology.