Introduction: Primary hepatic amyloidosis (PHA) is a rare disorder characterized by the deposition of insoluble protein fibrils in the liver. Clinical presentation is variable, ranging from asymptomatic to severe liver dysfunction. We present a case of PHA in a patient with abdominal distention and painless jaundice.
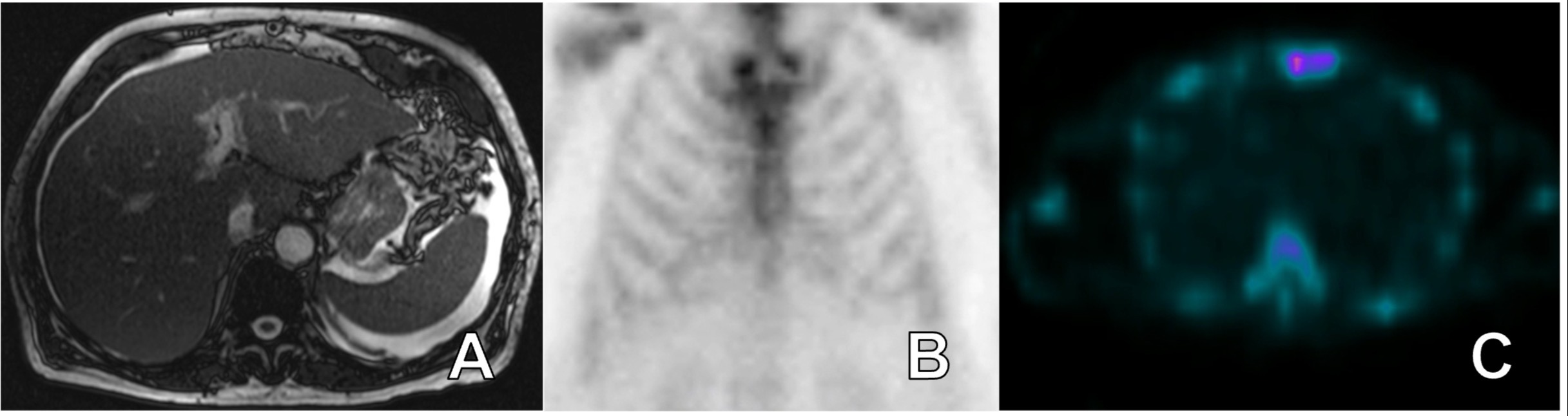
Case Description/Methods: 84-year-old male with a past medical history of coronary artery disease requiring coronary artery bypass presented to the hospital for several weeks of painless abdominal distention. Labs were remarkable for total bilirubin 6.5 mg/dL, alkaline phosphatase 482 U/L, AST 83 U/L, and ALT 47 U/L. Computed tomography abdomen pelvis with IV contrast showed moderate ascites with diffuse infiltration of the mesentery. There was no biliary obstruction, pancreatic mass, cirrhosis, or portal hypertension (PHT). Paracentesis revealed serum ascites albumin gradient > 1.1 and total protein < 2.5 consistent with sinusoidal PHT. Magnetic resonance imaging with cholangiopancreatography (MRI/MRCP) and upper endoscopy with endoscopic ultrasound revealed sequelae of PHT (A). Transjugular liver biopsy showed congophilic acellular material filling sinusoids and compressing hepatocytes consistent with amyloidosis. Amyloid protein identification showed kappa immunoglobulin light chains (AL). Cardiac MRI and hydroxymethylene diphosphonate (HMDP) ruled out cardiac amyloidosis (B). Bone marrow biopsy showed normocellular bone marrow. The patient was discharged on hospital day 17 and followed up with hematology amyloid specialist. He was started on a combination of Bortezomib, Cyclophosphamide, and Dexamethasone.
Discussion: Amyloidosis is considered a systemic disease, but specific organ involvement can occur in 10-20% of cases. AL amyloidosis is the most common type of amyloid in PHA. Abnormal protein folding and aggregation cause insoluble fibril deposition in hepatic parenchyma causing compression of hepatocytes and liver dysfunction. Clinical presentation is variable and non-specific including weight loss, fatigue, and abdominal discomfort. PHT and liver failure are rare complications. Diagnosis is based on liver biopsy with Congo red staining and immunohistochemistry. Treatment options are limited, and the prognosis depends on the underlying cause and the extent of liver damage. Median survival time is 10-14 months from diagnosis. Given the rarity of PHA and resultant poor outcomes, this case provides additional support for a multidisciplinary approach for the treatment of PHA.
Figure: Magnetic resonance imaging with cholangiopancreatography showed sequelae of portal hypertension and mild heterogenous hepatic parenchymal enhancement (A). Planar bone scintigraphy 99m-Technetium hydroxymethylene diphosphonate was negative for the presence of transthyretin cardiac amyloidosis. Heart to contralateral chest activity ratio was 0.92 (B). No detectable uptake of radiotracer by myocardium was appreciated on single-photon emission computed tomography (C).
Disclosures:
Sarah Grebennikov indicated no relevant financial relationships.
Vivek Jasti indicated no relevant financial relationships.
Ellen Tan indicated no relevant financial relationships.
Ruchi Bhatia indicated no relevant financial relationships.
Sarah Grebennikov, DO, MHS1, Vivek V. Jasti, MD2, Ellen Tan, DO1, Ruchi Bhatia, MD3. P2598 - Immunoglobulin Light Chain Amyloidosis With Hepatic Involvement Presenting as Painless Jaundice, ACG 2023 Annual Scientific Meeting Abstracts. Vancouver, BC, Canada: American College of Gastroenterology.