P3909 - Liver Re-Transplantation for Delayed Recurrence of Progressive Familial Intrahepatic Cholestasis Type 2 Phenotype After Living-donor Liver Transplantation
Digestive Disease Institute, Cleveland Clinic Abu Dhabi Abu Dhabi, Abu Dhabi, United Arab Emirates
Shiva Kumar, MD Digestive Disease Institute, Cleveland Clinic Abu Dhabi, Abu Dhabi, Abu Dhabi, United Arab Emirates
Introduction: Progressive Familial Intrahepatic Cholestasis (PFIC) is a heterogeneous group of rare genetic autosomal recessive diseases. Mutations in the ABCB11 gene, results in the absence of bile salt export pump (BSEP) in the canalicular membrane causing cholestasis and development of end-stage liver disease in the first decade of life in PFIC type 2. Recurrence of PFIC after liver transplant (LT) is extremely rare with almost all reported cases that required retransplant occurring following deceased donor LT (DDLT) in patients with PFIC type 2 with early recurrence. We present a rare case of late post-LT recurrent PFIC after living-donor LT (LDLT), resulting in graft failure necessitating re-transplant.
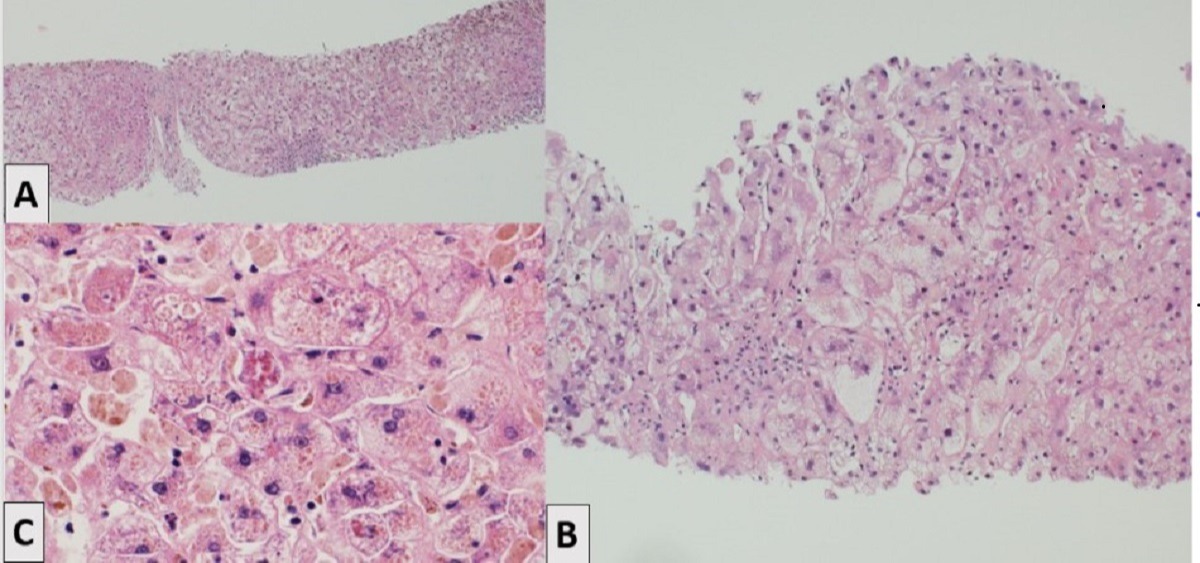
Case Description/Methods: A 17-year-old male with history of left lateral segment LDLT from his father at the age of 2 due to PFIC type 2 presented with increasing jaundice and pruritus after stopping immunosuppression for 2 years for unclear reasons. Initial laboratory tests revealed Bilirubin 17 mg/dL, ALT 287 U/L, Alkaline phosphatase 155 IU/L, AST 360 U/L, GGT 28 U/L. Liver biopsy revealed diffuse and severe hepatocellular and canalicular cholestasis with accompanying hepatocyte swelling and scattered giant cell transformation along with bile ductular reaction, without significant mixed inflammation, bile duct injury or endotheliitis, consistent with recurrent PFIC (Figure 1A, B). No clinical or biochemical improvement was noted with pulse steroids and escalation of immunosuppression, with continued clinical deterioration necessitating deceased donor liver re-transplantation. Examination of the explant confirmed findings consistent with recurrent PFIC (Figure 1C).
Discussion: Initially reported in 2009, recurrent PFIC is a rare entity, mostly reported following DDLT, resulting from antibody-mediated recurrent BSEP disease. Despite concerns related to LDLT in patients with an inheritable intrahepatic cholestatic disease, grafts from related donors do not appear to be at higher risk for failure from PFIC-related causes. As was the case in our patient, recurrence is limited to PFIC type 2 and is characterized by low-GGT cholestasis. The 15-year interval between primary LT to manifestation of recurrent PFIC and subsequent re-transplant is the longest reported in the literature, especially following LDLT. Since there is no proven therapy for antibody-mediated recurrent PFIC post LT, monitoring for the development of BSEP antibodies may enable early identification followed by appropriate intervention.
Figure: Hematoxylin and eosin (H&E) staining of liver core biopsy (A & B) and explanted liver (C)
Disclosures:
Shiva Kumar indicated no relevant financial relationships.
Shiva Kumar, MD. P3909 - Liver Re-Transplantation for Delayed Recurrence of Progressive Familial Intrahepatic Cholestasis Type 2 Phenotype After Living-donor Liver Transplantation, ACG 2023 Annual Scientific Meeting Abstracts. Vancouver, BC, Canada: American College of Gastroenterology.